(Look HERE for possible rotation projects!)
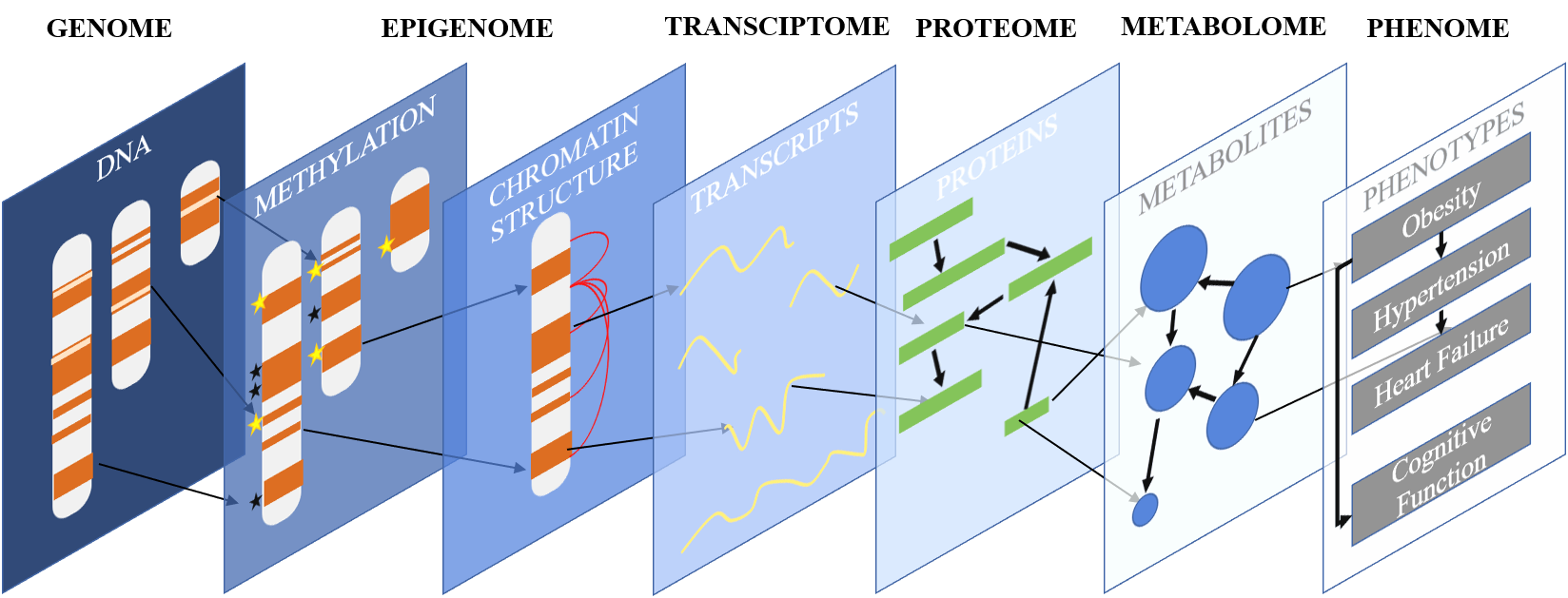
Our lab is focused on understanding how interactions between players at various biological scales result in phenotypic change. We study the association between DNA polymorphisms, CpG methylation, gene expression, cell-type-specific regulation, and clinical traits to gain a better understanding of how differences between individuals lead to dramatically different disease outcomes. We use a combination of bench and bioinformatic techniques to do so: our computational approaches inform our experimental work and our experimental work refines and improves our algorithm development.
Our work utilizes mouse genetic resource populations to study the progression of heart failure, a disease that is notoriously difficult to study using Systems Genetics approaches in human populations. Our primary resource populations are the Hybrid Mouse Diversity Panel (HMDP), containing over 150 fully inbred strains of mice, and the Collaborative Cross (CC), which contains as much genetic diversity as we see in human populations. These panels of mice show significant genetic and phenotypic diversity and have been used in labs around the world to study diseases ranging from atherosclerosis to bone density to hearing loss.
We have also recently begun working extensively with the Gene Tissue Expression (GTEx) cohort, a set of transcriptomes and DNA sequences from dozens of tissues drawn from the same group of 500+ individuals.
We combine our biological data with computational approaches, some from other labs, some home-grown, to understand how all of our high-throughput data combine together to create the clinical phenotypes we observe.
Our disease focus is Heart Failure, already the leading cause of hospitalization of people over the age of 65 and predicted to rise in incidence over the next decade even as overall rates of cardiovascular disease fall. The heart is a remarkably resilient organ, beating tens of millions of times a year. Over time, it accumulates damage but continues to function as normal. In this early stage of Heart Failure, called cardiac hypertrophy, it is invisible unless specifically looked for by tests requiring expensive equipment and trained technicians. In time, however, the heart reaches its biological limits. There are not enough functional cardiomyocytes left to pump the heart hard and fast enough to maintain cardiac output and symptomatic Heart Failure begins, characterized by fatigue, dizziness, edema, and, ultimately, death. Because it operates in silence for so long, it is difficult, at this end-stage state, to treat effectively. It is our goal to help identify simple, easy approaches – blood biomarkers, gene polymorphisms, or other simple-to-test qualities of an individual – that will allow doctors to diagnose Heart Failure early and lead to better outcomes for patients.
Current Projects
1) Genetic Drivers of Cellular Composition in the Heart in Healthy and Failing States
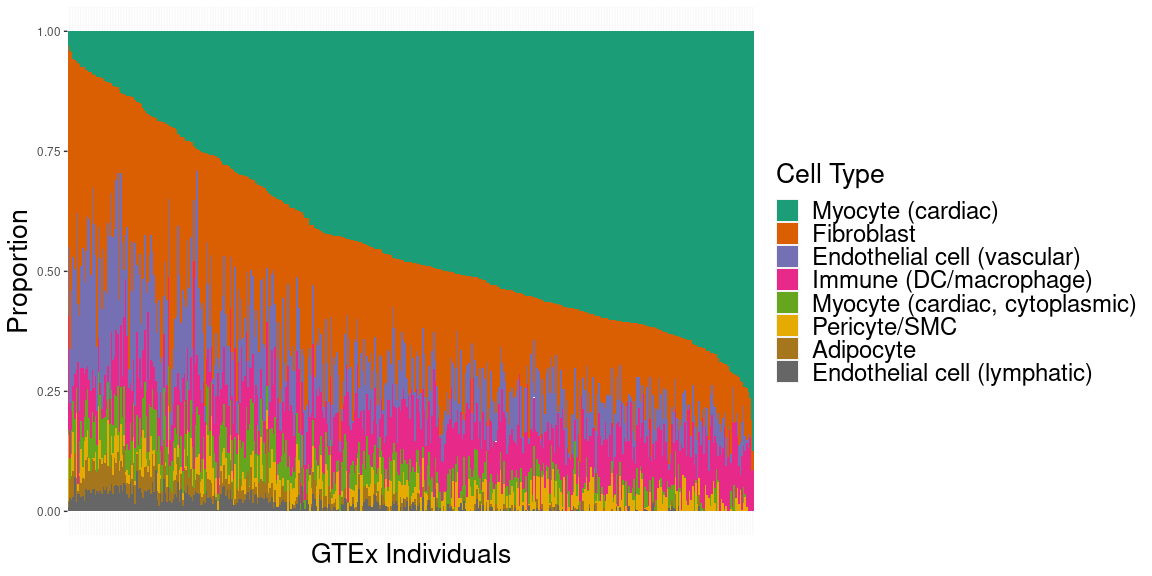
The underlying cellular composition of the heart is surprisingly poorly understood. Different groups, using different techniques and different model systems have reported wildly different proportions of cardiomyocytes to non-cardiomyocytes as well as different compositions of the non-cardiomyocyte component of the heart. Missing in these analyses is a study of how genetic backgrounds can lead to differences in cellular composition of heart tissue. We are using a combination of single-cell and single-nucleus datasets pulled from the heart, paired with population-scale bulk RNAseq datasets from the HMDP, CC, and GTEx populations to obtain estimates for cell-type composition in the heart across the entire HMDP and identify candidate genes and loci which lead to these changes.
2) Epigenetic Drivers of Heart Failure

Cardiac phenotypes are regulated by many underlying biological layers. One often overlooked layer is that of DNA methylation, which acts as an additional mediator of gene expression. Expanding on our prior work studying genetic polymorphisms in the HMDP cohort, we have performed Reduced Representational Bisulfite Sequencing in the HMDP both before and after catecholamine stimulation. We have identified a number of associations between the methylome and phenotypic traits in both the baseline and catecholamine-treated cohorts. Our research continues in identifying additional loci of interest and beginning the process of validation and mechanistic discovery using in vitro and in vivo models.
3) Function of Multinuclearity in Cardiomyocytes

A curious feature of mammalian cardiomyocytes is that the vast majority of them contain more than 2 copies of DNA within them. Pigs, for instance, have 2 or more nuclei present in over 95% of their cardiomyocytes. Prior work by us and collaborators have demonstrated that the percent of cells that are multinucleated across the HMDP strains are under genetic control. We identified a gene, Tnni3k, whose deletion affects multinucleation. Tnni3k was also identified by Doug Marchuk’s group at Duke as a driver of heart failure. Using a combination of single cell and spatial transcriptomics, we are trying to understand the links between multinucleation in cardiomyocytes and cardiac dysfunction.
4) Cross-Tissue Co-Expression Gene Networks
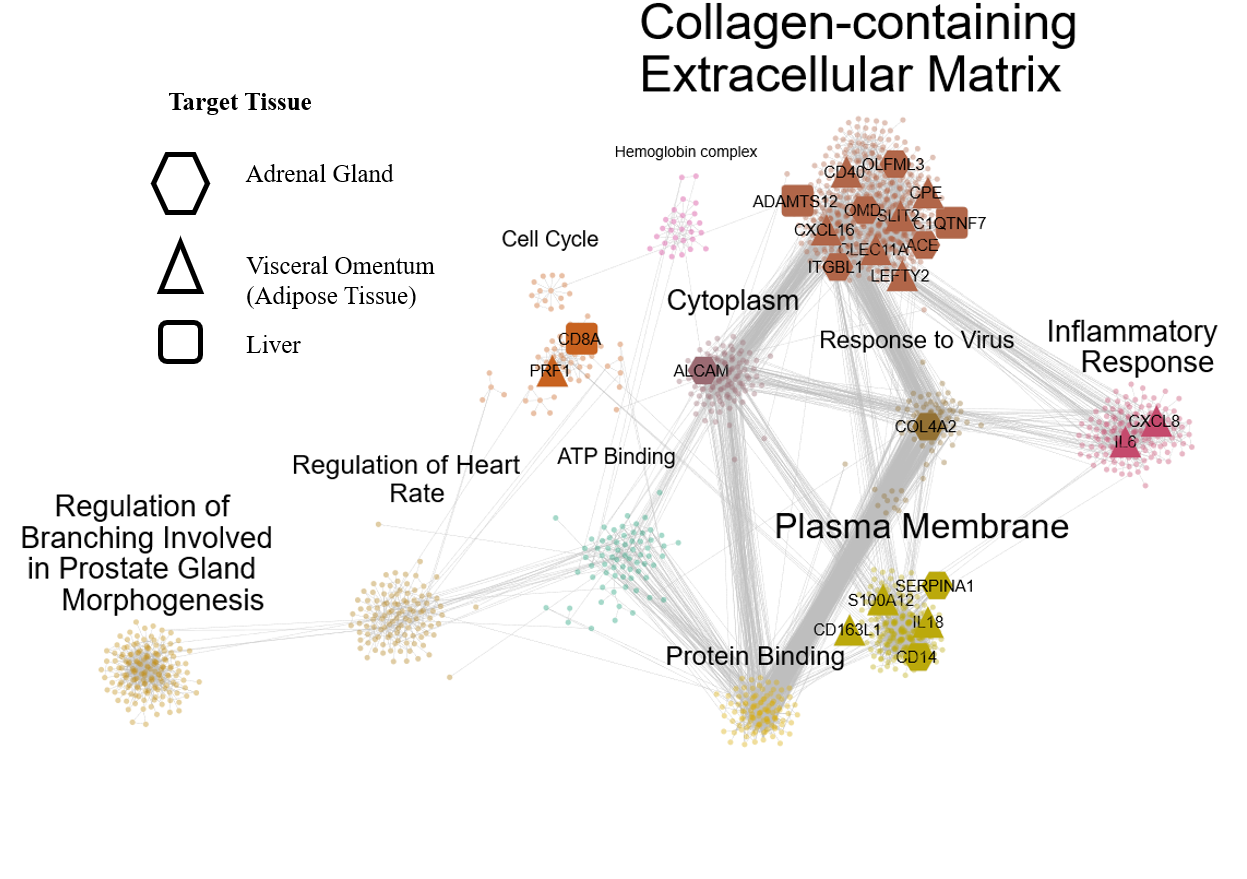
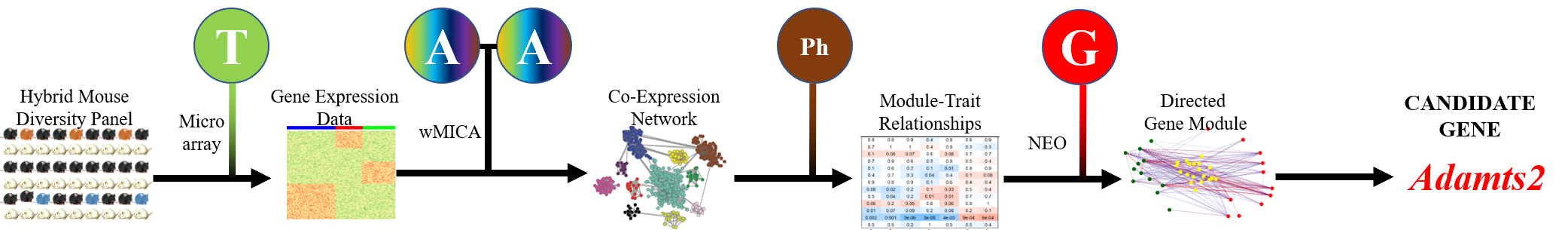
We have developed a new gene co-expression algorithm, weighted Maximal Information Component Analysis (wMICA) to explore transcriptomic co-expression networks in a way that allows for proportional membership within co-expression models and accounts for the non-linear relationships which are commonly observed between gene and protein abundances. This method has been used to identify novel regulators of cardiac phenotypes and as a hypothesis-generating tool for downstream validation.
We are now applying wMICA in a new way to data taken from both the CC and GTEx cohorts, merging it with the QENIE algorithm from the Seldin lab at UCI to identify cross-tissue gene networks linked together by secreted cytokines. Further research will allow us to better understand the ways that the entire body acts in a concerted fashion to maintain cardiac health and react to injury and stressors.